CRAM 3.1 becomes the default CRAM output version
Release 1.22 of HTSlib and SAMtools has switched to writing CRAM 3.1 by default. Read support CRAM 3.1 has been available since HTSlib 1.12 (March 2021), and we announced our switch to making it the default in September 2024.
Support for CRAM 3.1 exists in other tools, including the Noodles (Rust) and at the time of writing a GitHub pull-request exists for adding it to HTSJDK.
If you need to continue using CRAM 3.0, please explicitly specify the version when using SAMtools. For example:
samtools view -O cram,version=3.0 file.bam -o file.cram
This is also the way you can control the compression levels and/or profiles. E.g.
samtools view -O cram,small,level=7 file.bam -o file.cram
Note not all machine types may benefit from CRAM 3.1, as it largely depends on the randomness of the data. However some platforms may benefit considerably. As with CRAM 3.0 there is a tradeoff between speed and size, but CRAM 3.1 adds more sequence data specific compression codecs which make this tradeoff more worth while. The default output however favours the faster end of the speed/size tradeoff. There is a paper describing these codecs at https://doi.org/10.1093/bioinformatics/btac010.
CRAM 3.0 vs 3.1 benchmark summary
We compare CRAM 3.0 against CRAM 3.1 for a variety of sequencing platforms. Each line represents a different compression profile, targetting different positions on the speed-vs-size tradeoff.
Details of these compression profiles and specific tables of data are below. Some earlier benchmarks from 2019 are also available.
CRAM benchmarking profiles
(Updated Apr 2025)
These benchmarks here are for a variety of instrument types and showing differences between BAM and various CRAM versions. Each format also permits a variety of compression options and levels. For speed, we test only a subset of the full data sets. Note that this may have an impact on expected compression ratios if the data set contains a large amount of unaligned data at the end or if the chosen subset is not representative of the overall data.
The benchmarks below utilise the HTSlib supported compression profiles of “fast”, “normal” (the default), “small” and “archive”. These set a mix of options as defined here:
| Profile | CRAM versions | options |
|---|---|---|
| fast | 3.0 | seqs_per_slice=10000,level=1 |
| fast | 3.0, 3.1 | seqs_per_slice=10000,level=1,use_tok=0 |
| normal | 3.0, 3.1 | seqs_per_slice=10000 |
| small | 3.0 | seqs_per_slice=25000,level=6,use_bzip2 |
| small | 3.1 | seqs_per_slice=25000,level=6,use_bzip2,use_fqz |
| archive | 3.0 | seqs_per_slice=100000,level=7,use_bzip2 |
| archive | 3.1 | seqs_per_slice=100000,level=7,use_bzip2,use_fqz,use_arith |
If level 8 was specified prior to enabling “archive” mode, then it also adds “use_lzma” into the option list. We also provide benchmarks of using lzma to show the maximum compression, but it is rarely worth the CPU cost.
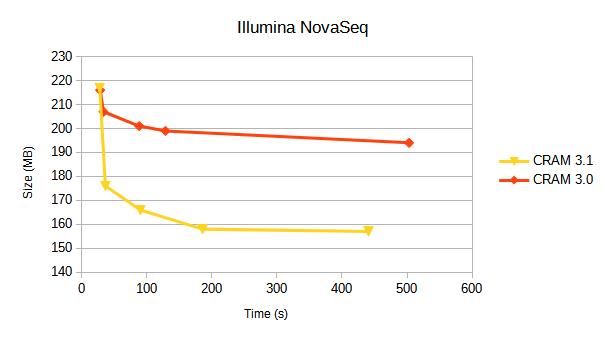
Illumina NovaSeq
10 million coordinate sorted alignments, originating from an Illumina published dataset when announcing the NovaSeq.
| Format | Options | Size(Mb) | Encoding(s) real | Encoding(s) CPU | Decoding(s) real | Decoding(s) CPU |
|---|---|---|---|---|---|---|
| BAM | level=1 | 577 | 5.6 | 18.3 | 0.8 | 4.4 |
| BAM | 515 | 9.2 | 61.7 | 0.8 | 4.2 | |
| BAM | level=7 | 508 | 15.0 | 109.9 | 0.9 | 4.3 |
| BAM | level=9 | 481 | 209.9 | 1661.0 | 0.9 | 4.3 |
| CRAM v3.0 | fast | 216 | 5.2 | 28.3 | 2.0 | 13.6 |
| CRAM v3.0 | 207 | 5.4 | 33.4 | 2.1 | 13.8 | |
| CRAM v3.0 | small | 201 | 11.6 | 88.3 | 3.2 | 24.4 |
| CRAM v3.0 | archive | 199 | 17.4 | 128.7 | 3,5 | 25.9 |
| CRAM v3.0 | archive,use_lzma | 194 | 65.4 | 503.2 | 2.6 | 18.6 |
| CRAM v3.1 | fast | 217 | 4.6 | 27.6 | 1.9 | 10.9 |
| CRAM v3.1 | 176 | 5.4 | 36.4 | 1.9 | 11.6 | |
| CRAM v3.1 | small | 166 | 11.8 | 90.1 | 5.5 | 41.5 |
| CRAM v3.1 | archive | 158 | 24.3 | 185.8 | 5.8 | 42.4 |
| CRAM v3.1 | archive,use_lzma | 157 | 57.2 | 440.9 | 6.0 | 43.4 |
A break down of data types in CRAM 3.1 is:
| Data type | File percentage | Bits per base |
|---|---|---|
| Quality | 50% | 0.46 |
| Sequence | 23% | 0.22 |
| Read names | 18% | 0.17 |
| Aux tags | 9% | 0.08 |
The quantisation of quality values shows a significant reduction in the amount of storage taken by quality values compared to the earlier HiSeq (below).
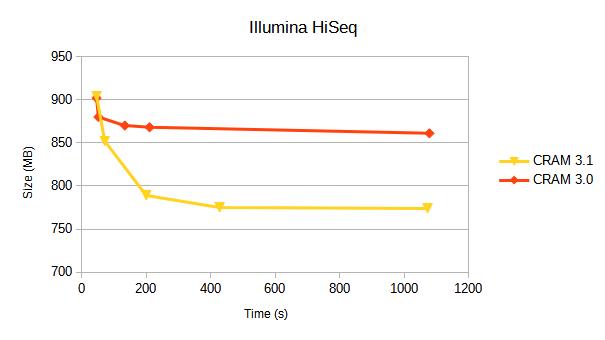
Illumina HiSeq 2500
10 million alignments from https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/HG002_NA24385_son/NIST_Illumina_2x250bps
Aligned and coordinate sorted.
| Format | Options | Size(Mb) | Encoding(s) real | Encoding(s) CPU | Decoding(s) real | Decoding(s) CPU |
|---|---|---|---|---|---|---|
| BAM | level=1 | 1671 | 12.6 | 43.7 | 1.8 | 10.4 |
| BAM | 1546 | 19.2 | 125.2 | 1.7 | 9.9 | |
| BAM | level=7 | 1536 | 28.9 | 204.4 | 1.7 | 9.9 |
| BAM | level=9 | 1461 | 251.4 | 1992.1 | 1.8 | 9.9 |
| CRAM v3.0 | fast | 902 | 7.7 | 45.9 | 3.1 | 21.2 |
| CRAM v3.0 | 880 | 8.1 | 51.6 | 3.0 | 21.5 | |
| CRAM v3.0 | small | 870 | 17.8 | 133.6 | 4.3 | 31.4 |
| CRAM v3.0 | archive | 868 | 28.1 | 210.4 | 4.5 | 32.6 |
| CRAM v3.0 | archive,use_lzma | 861 | 137.7 | 1078.5 | 3.9 | 27.3 |
| CRAM v3.1 | fast | 904 | 7.7 | 46.0 | 2.9 | 15.2 |
| CRAM v3.1 | 852 | 10.1 | 71.4 | 3.1 | 22.2 | |
| CRAM v3.1 | small | 789 | 25.9 | 200.0 | 14.6 | 113.3 |
| CRAM v3.1 | archive | 775 | 56.4 | 427.7 | 15.7 | 109.4 |
| CRAM v3.1 | archive,use_lzma | 774 | 135.1 | 1073.0 | 15.7 | 109.4 |
The addition of lzma as a codec choice helps the compression ratio of CRAM 3.0 a little, but at a big cost in CPU making it likely not worth it. It’s used even less in CRAM 3.1, costing CPU during encoder evaluation, but mostly being unutilised for any large data types thanks to the better range of codecs available.
A break down of data types in CRAM 3.1 (default) is:
| Data type | File percentage | Bits per base |
|---|---|---|
| Quality | 86% | 2.37 |
| Sequence | 7% | 0.18 |
| Read names | 4% | 0.11 |
| Aux tags | 3% | 0.08 |
The bulk of the storage cost is the quality values due to the 32 discrete values.
The QS data series shrinks from 735MB to 665MB when we enable archive mode, and this accounts for the bulk of the reduction. (Similarly for small mode which also uses the fqzcomp quality codec.)
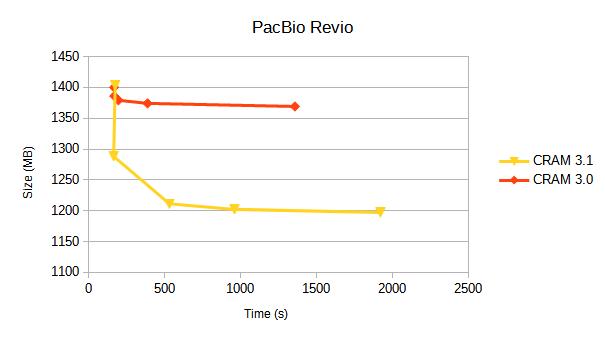
PacBio Revio
Alignments from https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=T2T/HG002/assemblies/polishing/HG002/v1.0/mapping/hifi_revio_pbmay24/
The reference used for this is the T2T hg002v1.0.1.fasta.gz file.
| Format | Options | Size(Mb) | Encoding(s) real | Encoding(s) CPU | Decoding(s) real | Decoding(s) CPU |
|---|---|---|---|---|---|---|
| BAM | level=1 | 5347 | 39 | 154 | 6.7 | 46.4 |
| BAM | 4832 | 66 | 420 | 5.6 | 32.3 | |
| BAM | level=7 | 4785 | 108 | 753 | 6.7 | 35.7 |
| BAM | level=9 | 4409 | 1219 | 9680 | 6.9 | 36.3 |
| CRAM v3.0 | fast | 1400 | 28 | 167 | 12.4 | 80.2 |
| CRAM v3.0 | 1386 | 27 | 168 | 12.1 | 77.3 | |
| CRAM v3.0 | small | 1379 | 29 | 195 | 12.1 | 79.9 |
| CRAM v3.0 | archive | 1374 | 63 | 387 | 16.6 | 107.8 |
| CRAM v3.0 | archive,use_lzma | 1369 | 248 | 1357 | 16.9 | 109.6 |
| CRAM v3.1 | fast | 1403 | 31 | 174 | 10.9 | 48.8 |
| CRAM v3.1 | 1288 | 27 | 164 | 10.7 | 44.1 | |
| CRAM v3.1 | small | 1211 | 69 | 531 | 37.6 | 288.6 |
| CRAM v3.1 | archive | 1202 | 144 | 959 | 39.4 | 298.7 |
| CRAM v3.1 | archive,use_lzma | 1197 | 310 | 1920 | 39.9 | 301.8 |
A break down of data types in CRAM 3.1 is:
| Data type | File percentage | Bits per base |
|---|---|---|
| Quality | 80% | 0.67 |
| MM+ML tags | 14% | 0.12 |
| Sequence | 3% | 0.03 |
| Read names | <1% | 0.00 |
| Other | 2% | 0.02 |
The quantised quality values compress almost as well as NovaSeq, but they make up a larger proportion of data partly due to the extreme compression of sequence values. To some degree this is an artifact of this specific data set which has been aligned against a diploid T2T assembly rather than the canonical GRCh38, but will also be in part due to the high quality of HiFi base calls.
The base modification tags are taking up a significant proportion and the ML confidence values could benefit from a similar quantisation seen in the quality scores.
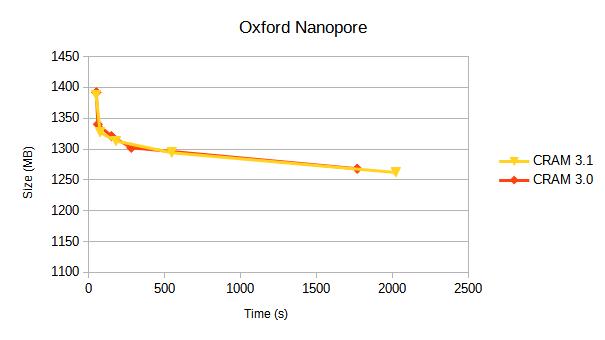
Oxford Nanopore
This is a the latest GIAB data uses a modern chemistry and base caller. The data comes from https://epi2me.nanoporetech.com/giab-2025.01/.
The data is approx 110,000 reads, from chr4:20M-50M.
| Format | Options | Size(Mb) | Encoding(s) real | Encoding(s) CPU | Decoding(s) real | Decoding(s) CPU |
|---|---|---|---|---|---|---|
| BAM | level=1 | 2347 | 17 | 55 | 1.8 | 11.9 |
| BAM | 2110 | 21 | 129 | 2.0 | 11.8 | |
| BAM | level=7 | 2100 | 38 | 207 | 2.2 | 12.5 |
| BAM | level=9 | 1990 | 189 | 1434 | 2.2 | 12.7 |
| CRAM v3.0 | fast | 1392 | 9 | 50 | 3.9 | 24.6 |
| CRAM v3.0 | 1340 | 10 | 60 | 3.8 | 24.0 | |
| CRAM v3.0 | small | 1321 | 22 | 149 | 4.3 | 28.7 |
| CRAM v3.0 | archive | 1302 | 39 | 280 | 4.1 | 28.4 |
| CRAM v3.0 | archive,use_lzma | 1268 | 226 | 1767 | 5.2 | 36.6 |
| CRAM v3.1 | fast | 1388 | 8 | 48 | 3.1 | 16.4 |
| CRAM v3.1 | 1327 | 11 | 75 | 3.6 | 22.2 | |
| CRAM v3.1 | small | 1313 | 29 | 179 | 3.6 | 23.4 |
| CRAM v3.1 | archive | 1294 | 72 | 546 | 11.0 | 83.6 |
| CRAM v3.1 | archive,use_lzma | 1262 | 258 | 2021 | 10.1 | 75.3 |
A break down of data types in CRAM 3.1 is:
| Data type | File percentage | Bits per base |
|---|---|---|
| Quality | 50% | 2.53 |
| MM+ML tags | 44% | 2.22 |
| Sequence | 6% | 0.30 |
| Read names | <1% | 0.01 |
| Other | <1% | 0.00 |
Base qualities are more costly to store than any other technology, mainly due to the high variability and no quantisation.
Base modifications are a surprising total of the compressed data, possibly due to the number of base types present, but also as with Revio there is no quantisation of the ML tag modification likelihoods.
The unpredictability of both of these data types means both CRAM 3.0 and 3.1 struggle to get effective compression.